GBA1基因编码一种溶酶体酶,可将葡萄糖神经酰胺 (GlcCer) 水解为葡萄糖和神经酰胺,GBA1基因的隐性遗传突变是导致戈谢病发生的原因,戈谢病是一种溶酶体贮积病,属于罕见病的一种。在该疾病中,GBA1基因的缺失导致溶酶体功能障碍和GlcCer水平升高。来自美国贝勒医学院的研究人员发现,GBA1基因同源物Gba1b在神经胶质细胞中表达,但在神经元中不表达,其缺失介导的大量GlcCer通过外泌体从神经元运输到神经胶质细胞进而行使相关功能。相关成果以“Neuronal activity induces glucosylceramide that is secreted via exosomes for lysosomal degradation in glia”为题在今年7月的SCIENCE ADVANCES杂志上出版发表。
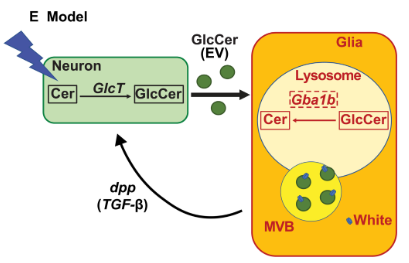
图示该研究的模式图。GlcCer在神经元活动后在神经元中合成,然后通过外泌体将其转运至神经胶质细胞,由Gba1b进行溶酶体降解。胶质细胞分泌的TGF-β/Dpp足以刺激神经元释放富含GlcCer的外泌体。White蛋白ABCG在GlcCer降解中的神经胶质细胞中起主要作用,并且与 MVBs 相关,而White蛋白和Gb1b的缺失协同并加剧了GlcCer的积累,表明两者独立参与GlcCer的降解。
葡萄糖神经酰胺Beta (GBA1) 基因的常染色体隐性突变是导致戈谢病 (GD)的原因,戈谢病是一种最常见的溶酶体贮积病。GBA1编码溶酶体水解酶 β-葡萄糖神经酰胺酶,可将葡萄糖神经酰胺 (GlcCer) 水解为葡萄糖和神经酰胺。GlcCer和半乳糖神经酰胺都属于鞘糖脂,绝大多数这些脂质的精确生物学功能尚不明确,但在严重儿童疾病的患者中发现了几种鞘糖脂的失调。GBA1基因的缺失可出现在婴儿中,导致严重的神经系统症状和两岁之前的死亡(II 型 GD)。许多与GD相关的GBA1突变与帕金森病 (PD)。这是由于神经酰胺和鞘磷脂的失衡会影响突触前末端的囊泡融合、运输和突触传递,神经酰胺水平的升高或降低通过调节膜流动性来影响神经元稳态。这些发现表明鞘糖脂代谢与罕见和常见的神经系统疾病之间存在密切关系。
GBA1的缺陷造成了神经细胞功能障碍及GD发病。在果蝇、鱼和小鼠中的研究表明,GBA1直系同源物的缺失会导致GlcCer积累、溶酶体功能障碍、泛素化蛋白质聚集、线粒体功能障碍、多巴胺能神经元丧失、进行性运动缺陷,寿命较短。然而,尽管GlcCer水平升高与神经退行性变之间存在关联,但许多关键问题仍未得到解答。这些包括细胞水平上GlcCer 产生和降解的来源和动态。GlcCer在GD病理生理学中的确切作用也不完全清楚。
对于GBA1的功能,果蝇中有两种人类GBA1的直系同源物:GBA1a和Gba1b。来自 modENCODE和 FlyAtlas的表达数据表明,Gba1a 主要在中肠道中表达,而Gba1b在大脑中表达。在本研究中,研究人员特别关注了Gb1b,通过使用CRISPR-Cas9插入Gal4 生成了果蝇直系同源物Gba1b的无效等位基因,建立了基因缺陷模型。结果发现,Gba1b在神经胶质细胞中表达并且行使重要功能,但在神经元中不表达,人GBA1基因的神经胶质特异性表达可以逆转果蝇Gba1b的全身性缺失造成的功能障碍。研究人员还证明,神经元活动触发神经元中GlcCer的合成,然后通过外泌体从神经元分泌。从神经元释放的外泌体GlcCer会被神经胶质细胞吸收,进行了溶酶体降解。在色素胶质细胞中大量表达的ABCG转运蛋白的White蛋白编码基因缺失,严重影响 GlcCer 的胶质溶酶体降解。最后,研究人员提出,GlcCer 通过外泌体从神经元转运到胶质细胞的机制在脊椎动物神经元细胞中是保守的,并且取决于胶质细胞中产生的转化生长因子-β (TGF-β)/骨形态发生蛋白 (BMP) 信号,而 ABCG转运蛋白White蛋白促进GlcCer转运到胶质细胞溶酶体进行降解。
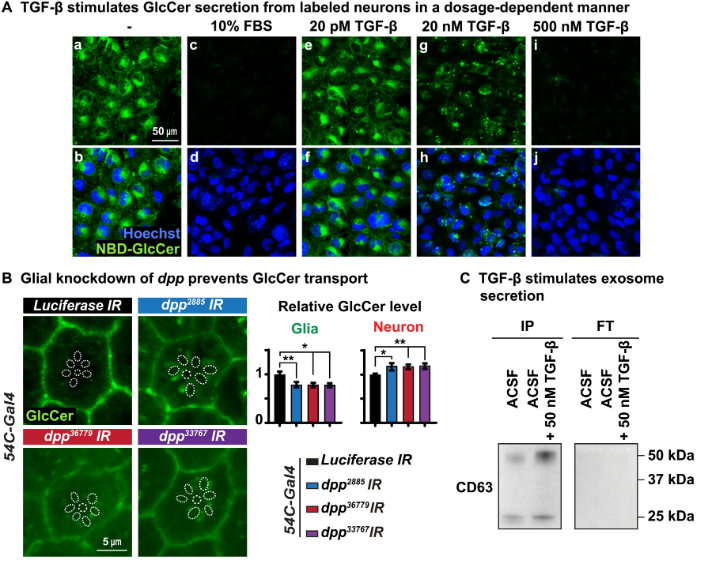
图:TGF-β促进GlcCer经外泌体传播和分泌。(A) 生长因子TGF-β刺激标记神经元NBD-GlcCer的释放。(B) dpp在胶质细胞敲除导致感光神经元中GlcCer升高和色素胶质细胞中 GlcCer水平降低。(C) NBD-GlcCer标记的神经元与两种条件ACSF培养基一起孵育(ACSF和50 nM TGF-β的ACSF 1 小时)。与ACSF 相比,50 nM TGF-β的ACSF含有更多来自 NBD-GlcCer 标记的神经元的外泌体。参考文献:Neuronal activity induces glucosylceramide that is secreted via exosomes for lysosomal degradation in glia. Sci Adv. 2022;8(28):eabn3326.



