p53突变(mutp53)丧失抑癌特性,驱动恶性肿瘤,但目前仍不清楚mutp53如何驱动肿瘤转移。来自清华大学生命科学学院、抗肿瘤蛋白质药物国家工程实验室罗永章教授课题组的研究人员发现,mutp53通过Rab偶联蛋白(RCP)介导的Hsp90α外泌体分泌来驱动细胞侵袭和癌症转移。该研究发表于Cell Reports杂志上。
p53是人类肿瘤中最常见的肿瘤抑制因子之一。功能研究已阐明,野生型p53(WTp53)通过与p53反应元件结合并调节下游基因表达来降低癌变前细胞的增殖和存活。大多数报道的p53突变都破坏了其肿瘤抑制活性,甚至某些突变导致了严重的肿瘤恶性转变。例如,携带TP53错义突变的Li-Fraumeni综合征(LFS)患者比没有p53表达的患者明显更早发生癌症。与此观察结果一致,具有p53突变的基因工程小鼠(R172H和R270H突变,分别相当于人中的R175H和R273H)比其p53杂合子或无效配对更能产生出侵略性和转移性的肿瘤。
突变p53(mutp53)在肿瘤中特异稳定并会积累,这是肿瘤功能获得(GOF)的关键特征和先决条件,这使mutp53成为癌症治疗中极为有吸引力的靶标。与正常组织相比,Hsp90分子伴侣机制在肿瘤中普遍激活,通过阻止mutp53免受E3泛素连接酶(如Mdm2和CHIP)介导的降解而稳定。Hsp90功能的稳定抑制显著抑制mutp53水平和肿瘤生长,从而导致mutp53小鼠的生存期延长。然而,考虑到Hsp90在细胞稳态中的关键作用,在许多旨在评估Hsp90抑制剂与化学治疗剂联合使用的潜在临床效果的临床试验中,以细胞渗透剂靶向Hsp90的细胞内伴侣功能已被证明是有效的。另一方面,已经证明Hsp90α是癌细胞分泌的。研究团队已经证明,在肿瘤细胞系和患者中,肿瘤分泌的细胞外Hsp90α(eHsp90α)水平与肿瘤恶性程度呈正相关。临床研究表明,血浆Hsp90α是早期检测各种类型癌症极好的生物标志物。值得注意的是,eHsp90α可与细胞外基质(ECM)蛋白或细胞表面受体相互作用,从而提高细胞运动性并支持肿瘤转移,从而成为有希望的抗肿瘤靶标。例如,通过与基质金属蛋白酶2(MMP-2)相互作用,eHsp90α帮助MMP-2保持稳定并激活MMP-2降解ECM,从而使肿瘤细胞能够离开原发性肿瘤而形成转移灶。此外,eHsp90α通过与低密度脂蛋白受体相关蛋白(LRP-1)相互作用而增加细胞运动性。这种相互作用诱导了一些传播信号通路的激活以及上皮到间充质的转变过程。研究团队已经证明,在几种小鼠模型中,靶向eHsp90α的细胞不可渗透的抑制剂或中和抗体可显著抑制肿瘤转移。
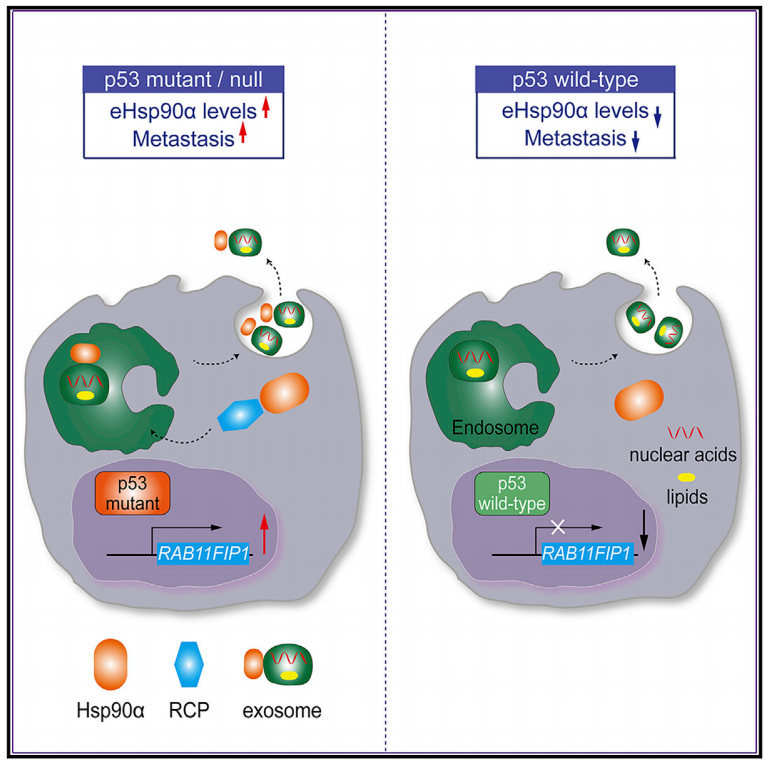
mutp53参与调节肿瘤分泌基因组,有利于肿瘤的侵袭和转移。Rab偶联蛋白(RCP)条件性基因敲除小鼠模型已用于mutp53驱动的胰腺癌转移中的研究。此外,RCP还参与了外泌体介导的肿瘤细胞与基质细胞之间mutp53的通讯。表达mutp53的肿瘤细胞的外泌体会在靶器官中引发转移前微环境,帮助转移定植。然而,从未研究过癌细胞中mutp53与Hsp90α分泌之间的关系。因此,特别是考虑到eHsp90α的促转移作用,研究人员因此研究了eHsp90α是否在细胞外环境中介导mutp53的促转移活性。
在这项研究里,研究人员发现WTp53抑制Hsp90α的分泌,而mutp53增强Hsp90α囊泡运输和外泌体介导的分泌。长期递送可以阻断细胞外Hsp90α(eHsp90α)功能的抗体,延长了p53-/-小鼠的生存期,并削弱了p53突变型肿瘤的侵袭性。此外,质谱和功能分析确定了RCP在mutp53诱导的Hsp90α分泌中的关键作用。RCP的抑制降低了eHsp90α水平并抑制肿瘤恶性进展。值得注意的是,重组Hsp90α的重新引入显著恢复了RCP缺少引起的迁移和侵袭能力受损。综上所述,这些发现阐明了mutp53通过其下游RCP介导的Hsp90α分泌执行致癌活性的分子机制,以及阐明了治疗mutp53高表达肿瘤的新策略。



