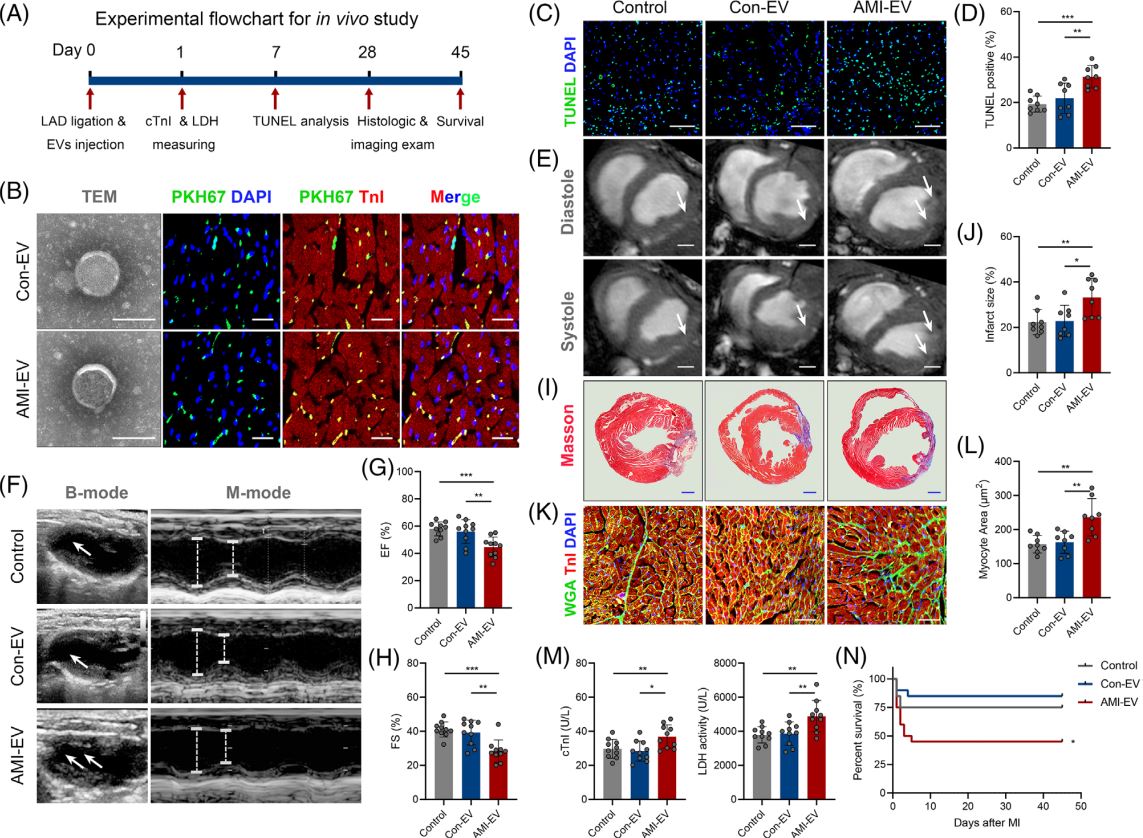
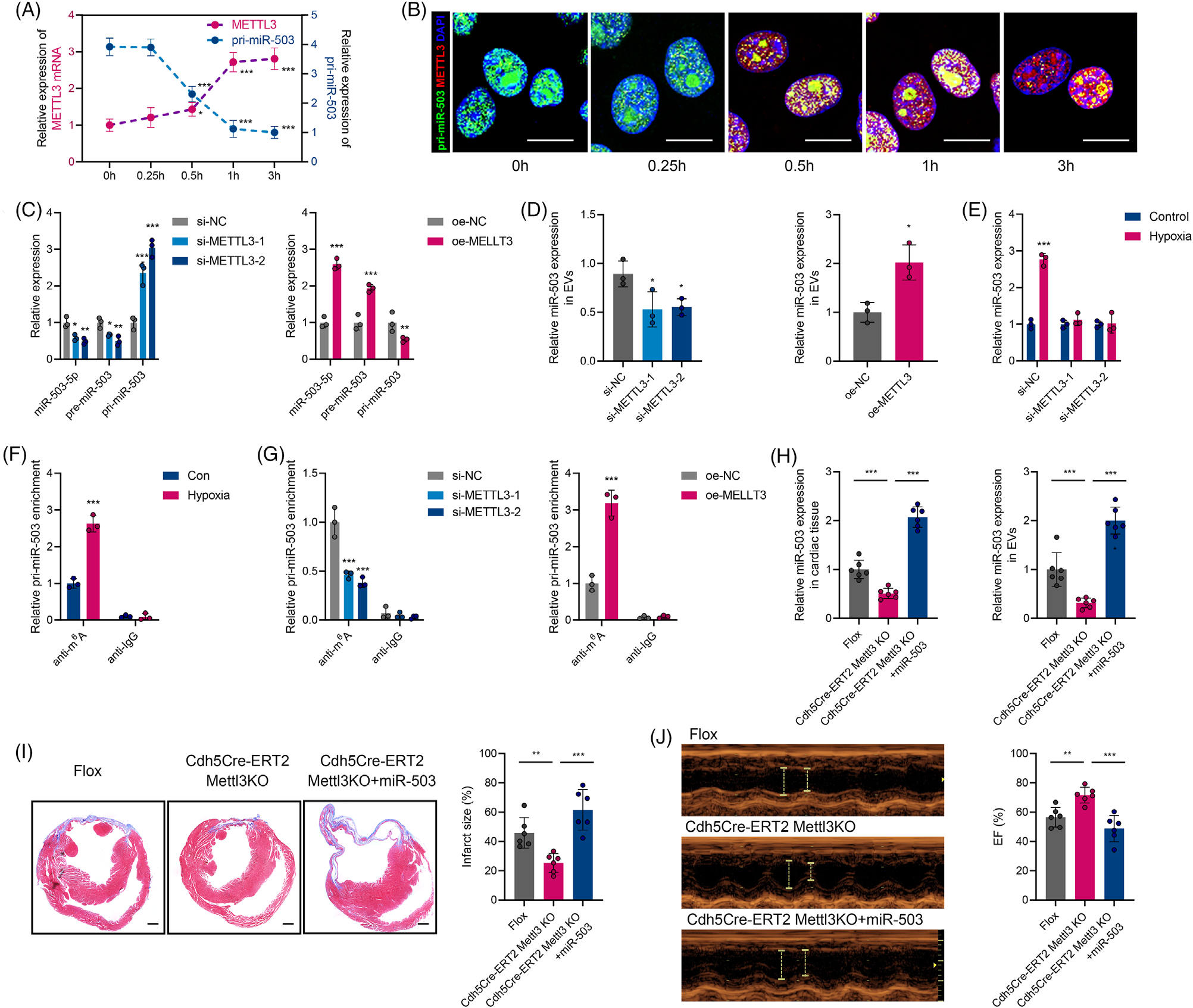
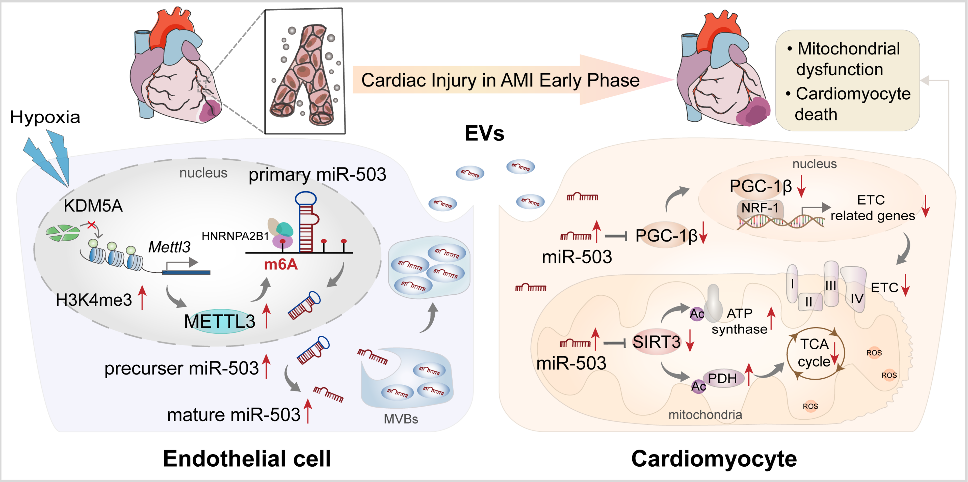
急性心肌梗死(AMI)是严重的致死性缺血性心脏病,表现为心肌局部缺血和不可逆的心肌细胞死亡。尽管近年来不断优化再灌注策略,急性期发生广泛组织损伤的患者仍表现出高死亡率,并可能进一步发展为心力衰竭。AMI早期心肌细胞损失的程度与预后直接相关。在AMI发病过程早期出现线粒体功能障碍和能量耗竭,而这严重危及心肌细胞存活。然而,AMI期间线粒体功能紊乱和心肌细胞丢失的机制亟待阐明。在AMI应激条件下,不同细胞群之间的通讯被广泛激活。越来越多的研究强调细胞外囊泡(EV)是心脏病理过程中生物信号的有效传递者。虽然已证实EV介导的细胞间通讯发生在心脏内和远距离器官中,但EV对AMI后心肌损伤的调控作用仍不明确。近日,哈尔滨医科大学附属第二医院张毛毛/田家玮教授在Clinical and TranslationalMedicine (IF:11.492) 上发表了题为“Extracellular vesicle-packaged mitochondrial disturbing miRNAexacerbates cardiac injury during acute myocardial infarction”的研究 (2022 Apr;12(4):e779),揭示了EV介导AMI后心肌损伤信号传递的重要作用。在该研究中,研究人员发现AMI诱导的EV可损害心肌细胞存活并加剧心脏损伤。EV包裹的miR-503在AMI早期富集,是介导心肌损伤的关键分子。功能研究显示,miR-503是通过靶向PGC-1β/NRF-1和SIRT3/PDH/ATP合酶通信轴触发线粒体功能障碍和心肌细胞死亡。在机制方面,确定了内皮细胞是AMI后miR-503的主要来源,缺氧快速诱导METTL3启动子区域H3K4me3,导致METTL3过表达,进而引起内皮细胞中m6A依赖的miR-503成熟。图1 急性心肌梗死诱导的细胞外囊泡加剧心肌损伤及新功能失调图2 miR-503通过靶向PGC-1β/NRF-1和SIRT3/PDH/ATP合酶通信轴触发线粒体功能障碍和心肌细胞死亡图3 在缺氧条件下内皮细胞METTL3可通过m6A介导pri-miR-503加工成熟总之,该研究提出了一种全新的AMI早期内源性损伤机制,即EV通过内皮细胞分泌的miR-503的穿梭加剧心肌损伤。干预miR-503的生物合成或使用miR-503拮抗剂可能是阻断EV介导的AMI后病理性细胞间通讯的有效手段,为AMI后早期心脏保护提供了全新的思路和干预靶点。哈尔滨医科大学附属第二医院孙萍博士为本文第一作者,张毛毛教授及田家玮教授为共同通讯作者。该研究得到了国家自然科学基金委、教育部心肌缺血重点实验室及黑龙江省博士后科学基金的资助。Extracellular vesicle-packagedmitochondrial disturbing miRNA exacerbates cardiac injury during acutemyocardial infarction. Clin Transl Med, 2022, 12: e779.外泌体资讯网 Clin Transl Med | 哈医大附属第二医院张毛毛/田家玮教授:细胞外囊泡携带线粒体扰乱的miRNA加剧心梗后心肌损伤