小型细胞外囊泡(EVs)是通过传递其分子来调节组织间通信的信号传递介质。近日,Nature Metabolism杂志上发表一篇文章,报道了肝脏来源的EVs是小鼠全身糖代谢控制的急性调节因子。肝脏EV分泌进入循环对高血糖作出反应,通过直接的器官间EV信号传递分别增加葡萄糖效应和胰岛素分泌,从而降低急性血糖。

非酒精性脂肪肝病(NAFLD)是肥胖的常见并发症,包括一系列肝脏疾病,非酒精性脂肪肝(NAFL,肝脏脂肪积累>5%)和更进一步的非酒精性脂肪性肝炎(NASH),后者的组织学定义为脂肪变性、小叶炎症和肝细胞肿胀。NAFLD与胰岛素抵抗、血脂异常以及2型糖尿病(T2D)和冠心病的风险密切相关;进展到NASH会增加纤维化、肝硬化、肝功能失调和与肝脏有关的死亡的风险。据估计,全球约有25%的人口患有NAFLD,其中约20%的NAFLD病例被归类为NASH。令人惊讶的是,肥胖者中NAFLD的发病率约为75%,而T2D患者中为56%。
NAFLD与糖尿病之间的密切关系的因素尚未完全理解,可能涉及多个贡献因素,包括肝脏胰岛素抵抗和低密度脂蛋白分泌的增加。NAFLD还与肥胖患者外周组织中胰岛素抵抗的发展相关,表明肝脏分泌的体液因子可能会影响远离胰岛素调节组织的胰岛素作用。事实上,被称为肝源性蛋白的肝脏分泌物的分泌在NAFLD中发生了变化,许多肝源性蛋白通过自分泌/旁分泌和内分泌信号传导途径诱导脂质代谢、外周胰岛素作用和血糖控制的变化。这些肝源性蛋白通过经典分泌从肝脏释放出来,这是一种含有N末端信号肽的蛋白质在内质网内被糖基化,并在涂有复合物II的涂层蛋白囊泡内被运送到高尔基体,然后融合并释放蛋白质到细胞膜外环境,以向靶细胞发出信号。虽然经典蛋白质分泌是组织通信的标志,但肝脏分泌的80-90%的蛋白质不含有信号肽,这表明通过其他机制分泌的蛋白质很可能是组织间通信和代谢控制的介质。
小型细胞外囊泡(EVs)是一种纳米级囊泡,其中包含脂质、蛋白质和核酸,可以转移到受体细胞并引发功能性反应。关于肝脏来源的EVs在调节其他细胞和组织中的过程以及EVs的组成如何随着NAFLD的进展而变化,我们知之甚少。一项研究表明,长期给予早期肥胖小鼠分离的肝细胞来源的EVs可以促进受体小鼠的胰岛素敏感性,这些效应被归因于单个微小RNA miR-3075的作用。相反,来自慢性肥胖小鼠的肝细胞EVs可能通过巨噬细胞的促炎激活导致胰岛素抵抗。虽然这项研究为了解肝细胞EVs的长期治疗如何促进组织适应以影响血糖控制奠定了重要基础,但目前尚不清楚肝实质其他细胞(例如库普弗细胞、肝星状细胞)的EVs是否影响了这些效应,这些反应在NAFL到早期NASH的进展过程中是否发生了变化,以及肝脏EVs是否激活了急性细胞信号事件并迅速调节了靶组织的代谢反应。此外,最近的研究表明,微小RNA(miRNA)是EVs的次要成分,携带miRNA的工程融合性EVs并未明显改变暴露于miRNA的细胞的功能性,这表明其他EV成分,如蛋白质,可能是调节EV介导通信的重要因素。鉴于EV的产生受激素和营养物质的影响,并且EVs携带多种细胞内和整合膜蛋白质,作者假设肝脏来源的EVs是对增加的营养物质可用性的反应,对受体细胞产生急性功能性变化以调节代谢。
该研究使用质谱蛋白质组学技术,识别了小鼠从健康肝脏到NAFL和NASH过程中肝脏来源的EVs蛋白质组成的变化。不论肝脏病理如何,来自小鼠和人类肝脏的EVs通过增加胰岛素分泌和改善葡萄糖效应(GE)来急性改善了血糖控制,与胰岛素敏感性的变化无关。这些反应特异于肝脏EVs,并且依赖于EV跨膜蛋白的存在。在健康状态下,肝脏EVs的分泌受到葡萄糖可用性的增强,而在胰岛素抵抗状态下受到抑制,表明肝脏来源的EVs在迅速调节血糖控制方面具有重要的生理作用,以应对波动的葡萄糖水平。‘
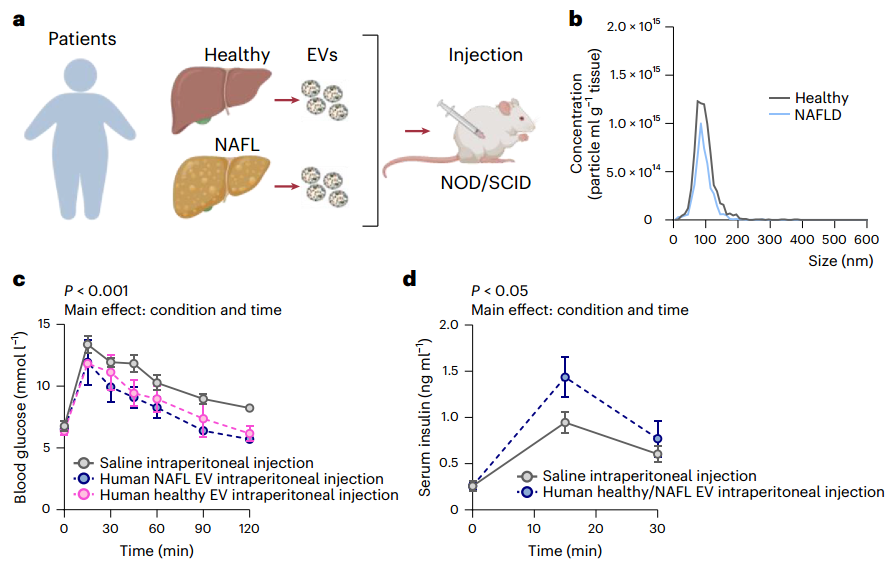
人类肝脏分泌的EV可改善血糖控制
尽管肥胖小鼠的肝脏来源EV蛋白质组发生了明显的重塑,但这种急性降血糖效应在健康和肥胖小鼠的非酒精性脂肪肝病中仍然存在。通过给予来源于有或无进行性非酒精性脂肪肝病的人类的肝脏EV,EV介导的降血糖效应得以重现,表明肝脏EV信号的广泛功能保守性和潜在治疗效用。总之,这项工作揭示了肝脏EV通过内分泌信号作用于外周组织以恢复餐后期的正常血糖水平的机制。
参考文献:
Miotto PM, Yang CH, Keenan SN, De Nardo W, Beddows CA, Fidelito G, Dodd GT, Parker BL, Hill AF, Burton PR, Loh K, Watt MJ. Liver-derived extracellular vesicles improve whole-body glycaemic control via inter-organ communication. Nat Metab. 2024 Jan 23. doi: 10.1038/s42255-023-00971-z. Epub ahead of print. PMID: 38263317.



