GBA1基因的变异会导致溶酶体葡萄糖脑苷酯酶(GCase)活性下降,是帕金森病(PD)和路易体痴呆(DLB)的常见风险因素。然而,GBA1突变的外显率并不完全(携带基因突变的人并不一定会表现出相关的疾病或症状),这表明还有其他基因参与了PD和DLB的发生机制。近日,美国西北大学的Dimitri Krainc研究组在Science杂志发表文章,该研究进行了全基因组CRISPR干扰筛选,鉴定出能够调节GCase活性的候选基因。其中,Commander复合体的一个成分COMMD3对于溶酶体GCase活性至关重要,其变异与帕金森病风险增加相关,这表明对该复合体的调控可能在决定GBA1介导的帕金森病风险中发挥作用。

GBA1基因的杂合致病性变异是帕金森病(PD)和路易体痴呆(DLB)的常见风险因素,而携带双等位基因致病性变异的人会发展成常染色体隐性遗传的溶酶体贮积病——戈谢病,并且也面临患PD的风险。GBA1编码溶酶体酶葡萄糖脑苷酯酶(GCase),其功能是水解葡萄糖脑苷脂。GBA1致病性变异会导致GCase活性降低,导致脂质底物(如葡萄糖脑苷脂和葡萄糖鞘氨醇)的积累。此外,即使没有GBA1致病性变异,溶酶体GCase活性下降也与线粒体功能障碍和α-突触核蛋白水平升高有关。严重降低GCase活性的致病性变异通常与更高的PD风险和更早的发病年龄相关。然而,由于GBA1变异在PD和DLB中的外显率并不完全,表明可能存在其他遗传因素修饰了GBA1携带者的疾病风险。研究表明,SNCA和CTSB位点附近的变异以及TMEM175基因中的特定变异与GCase活性下降及GBA1-PD风险增加有关。此外,相关研究发现罕见的溶酶体储存病相关基因变异在GBA1-PD患者中更为常见。为了更好地理解具体基因和细胞通路对GCase活性及溶酶体稳态的影响,研究团队进行了全基因组CRISPR干扰筛选,以鉴定GCase活性和溶酶体功能的遗传修饰因子。
全基因组CRISPRi筛选揭示溶酶体GCase活性的调控因子
研究通过流式细胞术结合底物PFB-FDGlu测量溶酶体GCase活性,并使用SCARB2敲除(KO)细胞作为阳性对照,验证了实验的准确性。随后,通过全基因组CRISPRi筛选鉴定了338个可能调控溶酶体GCase活性的基因,其中190个基因敲低导致GCase活性下降,148个基因敲低导致GCase活性增加。低活性组中,包括GBA1和SCARB2等已知基因,进一步验证了筛选的可靠性。前两位低活性基因为COMMD3和COMMD3-BMI1,后者是COMMD3与BMI1的转录产物;此外还包括COMMD9、RAB7A及HOPS复合体成员(VPS16、VPS39和VPS41)。高活性组中,IGF1R和REL位列前十。
小干扰RNA(siRNA)验证显示,SCARB2和VPS16敲低分别导致LIMP-2和VPS16蛋白减少80%和60%,并分别使GCase活性下降50%和40%。RAB7A、VPS16、VPS39和VPS41敲低后,GCase活性下降30%-60%。在携带致病性VPS16变异的患者成纤维细胞中,VPS16蛋白减少50%,GCase活性下降40%,进一步支持HOPS复合体基因的调控作用。
此外,针对高活性组基因IGF1R和REL,研究使用抑制剂Linsitinib和IT-603,发现两者均能显著提升HEK细胞中溶酶体GCase活性,且不影响底物摄取。IT-603还在携带GBA1突变的帕金森病患者分化神经元中提高了25%的GCase活性。本研究揭示了HOPS复合体及相关基因对溶酶体GCase活性的调控作用,为溶酶体功能障碍相关疾病提供了潜在的治疗靶点。
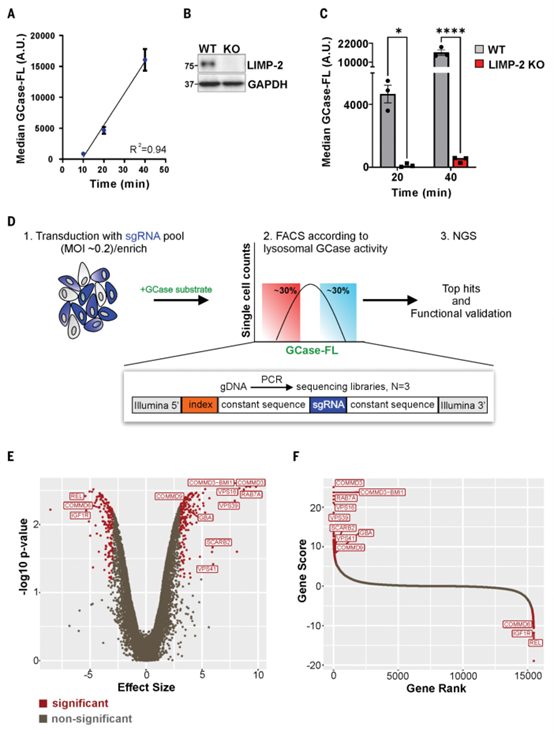
通过全基因组CRISPRi筛选鉴定溶酶体GCase活性的候选调控因子
COMMD3缺失降低了iPSC来源的小胶质细胞和神经元中的溶酶体GCase活性
COMMD3是由COMMD1至COMMD10、CCDC22和CCDC93组成的CCC复合体的一部分,并与Commander复合体共同参与内体回收。敲除COMMD3的HEK细胞显示溶酶体GCase活性降低50%-60%,且与底物摄取无关。在iPSC分化的小胶质细胞和神经元中,COMMD3缺失也显著降低了GCase活性。通过稳定表达COMMD3的HEK细胞实验,发现外源性COMMD3可恢复COMMD1的斑点形成、CCDC22表达及GCase活性。结果表明,COMMD3的缺失会破坏CCC和Commander复合体的功能,而重新引入COMMD3能够恢复溶酶体GCase活性,且该作用并非由BMI1介导。
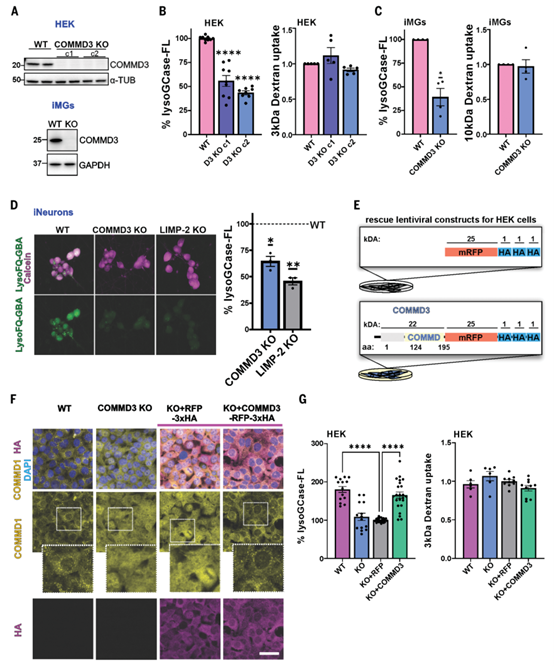
COMMD3的敲除降低了溶酶体GCase活性
COMMD3缺失损害溶酶体蛋白运输,导致溶酶体功能障碍
为探讨COMMD3敲除如何降低溶酶体GCase活性,研究分析了溶酶体相关蛋白的丰度。在COMMD3敲除(KO)和野生型(WT)HEK细胞全细胞裂解物中,GCase、LIMP-2、HEXB、LAMP2等溶酶体蛋白以及prosaposin、progranulin和cathepsin B的水平无显著差异。此外,透射电子显微镜观察和自动图像分析显示,WT和COMMD3-KO细胞的细胞器数量及LAMP1斑点密度相似。在COMMD3-KO的iPSC来源多巴胺能神经元中,GCase和LIMP-2的蛋白水平以及GBA1和SCARB2的基因表达也未发生变化。然而,DQ-BSA实验表明,COMMD3-KO细胞的溶酶体降解效率降低,尽管溶酶体酸度未显著改变。
通过LAMP1-RFP-3×HA标记的Lyso-IP纯化内溶酶体,发现COMMD3-KO细胞中LAMP2、LIMP-2、GCase和cathepsin B的水平较WT细胞分别降低30%-40%。在COMMD3敲除的iPSC来源多巴胺能神经元中,内溶酶体中上述蛋白水平均减少,表明COMMD3缺失阻碍了LIMP-2和GCase向内溶酶体的运输。此外,COMMD3-KO细胞中prosaposin水平升高至WT细胞的2.7倍,而saposin C(GCase的内源激活因子)的比例降低60%,显示prosaposin切割受损。同样,progranulin的全长蛋白浓度升高3倍,而其活性片段比例下降70%,表明progranulin加工受阻。
研究表明,CCC和Commander复合体的缺陷通过影响prosaposin和progranulin的加工,导致溶酶体GCase活性下降,并可能对溶酶体稳态产生更广泛的影响。
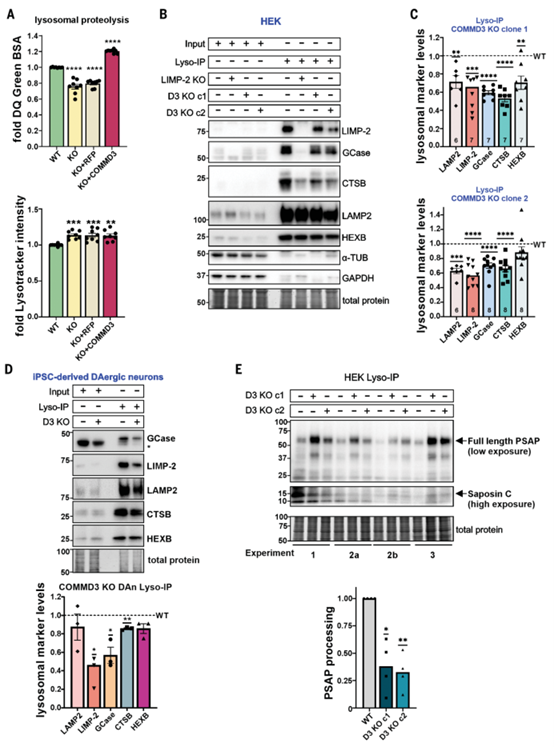
COMMD3功能的丧失导致溶酶体功能障碍
COMMD3缺失通过胞外囊泡释放溶酶体蛋白
研究发现,COMMD3缺失影响LIMP-2和GCase的内体运输。在RUSH实验中,LIMP-2和GCase在COMMD3敲除(KO)细胞中从高尔基体转运后,积累于早期内体标志物EEA1阳性的囊泡内,并最终通过胞外囊泡(EVs)释放。与野生型(WT)相比,COMMD3-KO细胞的EVs中LAMP1、LIMP-2和HEXB显著增加,HEXB含量提高四倍,且EV标志物FLOT1增加2.4倍。此外,COMMD3-KO的iPSC来源多巴胺能神经元的EVs中LAMP1和HEXB分别增加12倍和3倍。研究表明,COMMD3缺失通过早期内体滞留,推动溶酶体蛋白经胞外囊泡释放,而非正常运输至溶酶体。
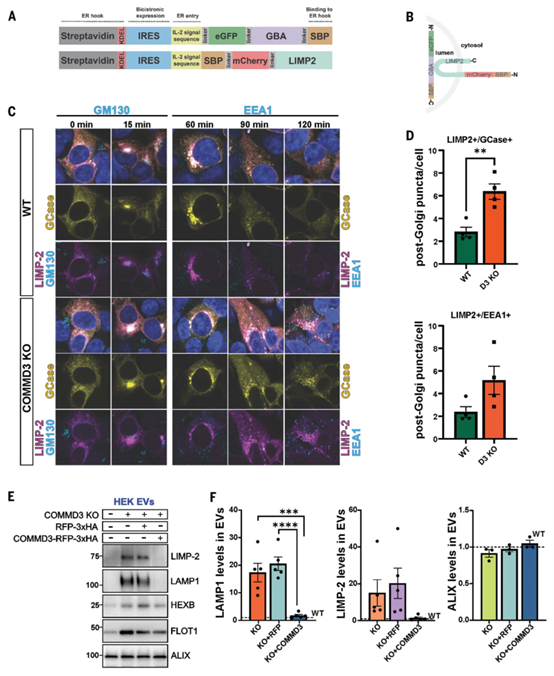
COMMD3缺失导致溶酶体蛋白滞留在高尔基体后的囊泡中,并通过胞外囊泡(EVs)释放
COMMD9缺失降低了iPSC来源神经元中的溶酶体GCase活性
COMMD9是COMMD蛋白家族中排名第二的关键基因。COMMD9敲除(KO)iPSCs被分化为神经元(iNeurons),MAP2和TUJ1染色显示,COMMD9或COMMD3的敲除并未影响神经元分化。COMMD9缺失导致COMMD1和CCDC22蛋白水平降低,并显著降低溶酶体GCase活性,验证了COMMD9作为GCase活性调控因子的作用。
CCC和Commander复合体基因中功能缺失变异的增加与PD风险增加相关
为探讨CCC和Commander复合体功能障碍与帕金森病(PD)风险的关系,研究对COMMD环(10个基因)、CCC复合体(13个基因)和Commander复合体(17个基因)中的罕见功能缺失变异(频率<1%)进行病例对照基因组负担分析。结合AMP-PD全基因组测序队列和UK Biobank全外显子组队列(共6166例PD病例和109,467名对照),发现COMMD环(P=1.79×10⁻⁵,OR=2.43)、CCC复合体(P=4.87×10⁻⁷,OR=2.56)和Commander复合体(P=1.79×10⁻⁵,OR=2.13)中罕见功能缺失变异在PD病例中显著富集。单基因分析中,COMMD9在UK Biobank队列中显著相关(FDR校正P=2.24×10⁻²,OR=6.10)。研究结果表明,Commander复合体及其组成部分中的罕见功能缺失变异与PD风险增加密切相关。
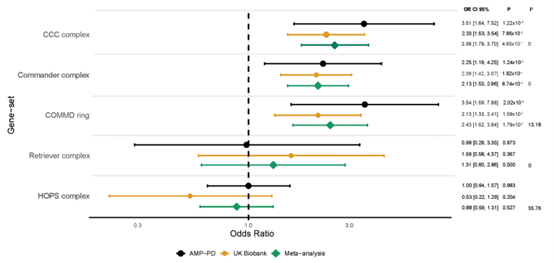
帕金森病中COMMD相关基因的功能缺失(LOF)变异负担增加
帕金森病或DLB患者前脑中Commander复合体蛋白的表达
研究分析了7例DLB患者和7例对照的前脑样本,发现DLB患者中COMMD3、COMMD1和CCDC22蛋白水平相较对照有所降低。然而,由于组内差异较大,需要更大样本量来明确WT CCC和Commander复合体缺陷在PD、DLB及其他神经退行性疾病中的潜在作用。
总之,该研究通过使用全基因组CRISPR干扰筛选,该研究鉴定出铜代谢MURR1结构域蛋白3(COMMD3),这是COMMD/卷曲螺旋结构域蛋白22(CCDC22)/CCDC93(CCC)和Commander复合体的一个成分,它是GCase和溶酶体活性的修饰因子。COMMD3的缺失会增加溶酶体蛋白通过细胞外囊泡的释放,导致这些蛋白无法有效传递到内溶酶体,从而引起溶酶体功能障碍。Commander基因家族中的罕见变异与帕金森病风险增加相关。因此,COMMD基因及其相关复合体能够调节溶酶体稳态,并可能成为与溶酶体功能障碍相关的帕金森病及其他神经退行性疾病的修饰因子。
Commander复合体由COMMD蛋白二聚体组成的异五聚环构成,其与Retriever复合体的稳定结合通过CCDC22和CCDC93的广泛相互作用介导。每个COMMD蛋白倾向于与另一个COMMD蛋白形成异二聚体,任何一个组分的丧失可能会破坏整个Commander复合体的稳定性。研究显示,COMMD3的缺失破坏了复合体的形成,而外源性COMMD3可恢复其稳定性。CCC和Commander复合体在将Retriever货物从内体回收到质膜中的作用至关重要,此外数据还揭示了它们在溶酶体功能中的扩展作用。
COMMD3缺失导致溶酶体蛋白部分滞留于内体中,随后通过胞外囊泡(EVs)释放,而非正常递送至溶酶体,最终引起溶酶体功能障碍。Commander复合体功能障碍还降低了溶酶体中GCase活性,部分原因是溶酶体内GCase及其转运蛋白LIMP-2减少,同时GCase或LIMP-2滞留于早期内体。此外,Commander功能的丧失通过抑制prosaposin向内源性GCase激活因子saposin C的转化,以及progranulin向granulin片段的转化,进一步降低GCase活性。这种progranulin转化障碍也可能通过影响溶酶体脂质BMP的稳定性,干扰溶酶体的整体稳态。此外,COMMD3功能丧失还与成熟的cathepsin B减少有关,而cathepsin B负责prosaposin和progranulin的加工,其减少可能是导致溶酶体功能障碍的重要途径,并与帕金森病(PD)风险相关。
研究还表明,COMMD基因家族中的罕见变异与PD风险增加相关。尽管此前未发现COMMD基因的常见或罕见变异与PD风险相关,但通过全基因组CRISPRi筛选和后续验证发现了这种关联。这些基因在质膜蛋白的转运中发挥重要作用,可能解释了其功能缺失变异在人群中的低频率。
研究结果表明,CCC和Commander复合体缺陷影响溶酶体稳态,COMMD变异可能是PD、DLB及其他具有溶酶体功能障碍的神经退行性疾病的风险调节因子。未来有必要研究针对COMMD通路的治疗是否可改善这些疾病中的溶酶体功能。
参考文献:
Minakaki G, Safren N, Bustos BI, Lubbe SJ, Mencacci NE, Krainc D. Commander complex regulates lysosomal function and is implicated in Parkinson's disease risk. Science. 2025 Apr 11;388(6743):204-211. doi: 10.1126/science.adq6650. Epub 2025 Apr 10. PMID: 40209002.



